




Содержание
Перейти к:
https://doi.org/10.17749/2070-4909/farmakoekonomika.2023.170
Перейти к:
На сегодняшний день сложно переоценить новые направления в фармакотерапии периферических Т-клеточных лимфом (ПТКЛ): иммунотерапии, в т.ч. адоптивной, таргетной и химиотерапии. Тем не менее биомаркеров, которые бы предсказывали ответ на лечение, крайне мало. Большую проблему составляют пациенты с рефрактерной и рецидивирующей ПТКЛ, которые не отвечают на подобную терапию или у них развиваются нежелательные явления, что делает актуальным вопрос персонификации лечения и поиска новых предиктивных маркеров с последующей тщательной аналитической и клинической валидацией в реальной врачебной практике. В литературе подчеркивается важность применения биомаркеров, полученных в результате полноэкзомного секвенирования и секвенирования транскриптома опухолей. В обзоре рассмотрен Т-клеточный онтогенез, а также возможности персонализации таких противоопухолевых препаратов, как азацитидин, дувелисиб, ромидепсин и бортезомиб, для терапии рефрактерной или рецидивирующей ПТКЛ.
Сорокина М.А., Рахтеенко А.В., Гришина Т.Р. Новые фармакотерапевтические подходы к лечению периферической Т-клеточной лимфомы. ФАРМАКОЭКОНОМИКА. Современная фармакоэкономика и фармакоэпидемиология. 2023;16(2):291-302. https://doi.org/10.17749/2070-4909/farmakoekonomika.2023.170
Sorokina M.А., Rakhteenko A.V., Grishina T.R. New pharmacotherapeutic approaches for the treatment of peripheral T-cell lymphoma. FARMAKOEKONOMIKA. Modern Pharmacoeconomics and Pharmacoepidemiology. 2023;16(2):291-302. (In Russ.) https://doi.org/10.17749/2070-4909/farmakoekonomika.2023.170
Периферические Т-клеточные лимфомы (ПТКЛ) – это подвид неходжскинских лимфом (НХЛ), происходящих из зрелых (посттимических) Т-лимфоцитов и NK-клеток. ПТКЛ является редкой нозологией (менее 1 случая на 100 тыс. населения в год), на долю которой приходится порядка 5–10% НХЛ [1].
В июне 2022 г. была пересмотрена классификация опухолей лимфоидной ткани Всемирной организации здравоохранения [2], согласно которой выделяют 9 вариантов зрелых Т/NK-клеточных неоплазий (классификация основана на различных подходах: клиническая картина, локализация заболевания, клетка-прекурсор / стадия дифференцировки или цитоморфология):
Внутри каждого варианта зрелых Т/NK-клеточных неоплазий, в свою очередь, содержится от 1 до 9 подтипов [2], что делает данную нозологию крайне гетерогенной и сложной в диагностическом плане. Самыми распространенными подтипами ПТКЛ являются ПТКЛн – 26%, ангиоиммунобластная Т-клеточная лимфома (АИТЛ) – 19%, ALK1-позитивная АККЛ – 12%, Т-клеточный лейкоз/лимфома взрослых – 10%, NK/T-клеточная лимфома – 10%, интестинальные формы ПТКЛ – 5%. Порядка 12% ПТКЛ реклассифицируются в другие типы ходжкинских и неходжкинских лимфом. Все остальные варианты встречаются крайне редко и в сумме составляют не более 6% от всех ПТКЛ [1].
По данным Международного проекта по Т-клеточным лимфомам, 5-летняя выживаемость при ПТКЛ составляет не более 32% для ПТКЛн, АИТЛ, NK/T-лимфомы по сравнению с 14% для Т-клеточного лейкоза/лимфомы взрослых [1]. Вне зависимости от подтипа ПТКЛ характер заболевания, за редкими исключениями, агрессивный (например, ALK-позитивная АККЛ), а прогноз неблагоприятный.
Для установления прогноза при НХЛ применяется международный прогностический индекс (англ. international prognostic index, IPI). IPI учитывает 4 независимых прогностических фактора [3]:
На основании этих показателей определяются группы с низкой, низкой/промежуточной, высокой/промежуточной и высокой степенями риска раннего прогрессирования.
Для оценки ответа на терапию при НХЛ применяются критерии Lugano: полный ответ (англ. complete response, CR), частичный ответ (англ. partial response, PR), стабильное заболевание (англ. stable disease, SD) и прогрессирование лимфомы (англ. progressive disease, PD) [4].
С целью персонализированного назначения фармакотерапии ПТКЛ необходимо выявить нарушения сигнальных каскадов, вовлеченных в процессы роста и дифференциации Т-клеток. В последние годы становится очевидным, что методы полноэкзомного секвенирования (англ. whole exome sequencing, WES) и секвенирования транскриптома (англ. RNA-sequencing, RNA-seq) позволяют находить информативные биомаркеры, способствующие пониманию патогенеза ПТКЛ у конкретного пациента, дифференциальной диагностике, прогнозу и стратификации пациентов в ответ на терапию [5]. При этом результаты, полученные с использованием данных WES и RNA-seq, существенно коррелируют с результатами иммуногистохимического анализа [6]. Заметим, что эффекты произвольных молекул на транскрипцию генов в различных типах клеток (в т.ч. опухолевых) могут успешно прогнозироваться с использованием методов хемотранскриптомного анализа, развиваемых в научной школе академика Ю.И. Журавлева [7–9].
Таким образом, разработка новых вариантов терапии ПТКЛ, а также поиск биомаркеров ответа и резистентности на новые виды терапии могут проводиться в рамках парадигмы постгеномных исследований, включая транскриптомику и хемотранскриптомное моделирование опухолевых клеток. Далее последовательно рассмотрены дифференцировка Т-клеток, а также известные в настоящее время данные о назначении азацитидина, дувелисиба, ромидепсина и бортезомиба для терапии рефрактерной или рецидивирующей ПТКЛ.
Большинство периферических Т-клеток происходят из лимфоидных предшественников, которые созревают в тимусе. Основные принципы созревания и дифференцировки Т-клеток важны для понимания биологии ПТКЛ.
Покинув костный мозг, ранние лимфоидные предшественники входят в кору тимуса в корково-медуллярном соединении. Этот процесс опосредован набором хемокинов и поверхностных рецепторов, включая CXCL12, CCL21, CCL25, CCR9 и CCR7. В коре тимуса ранние лимфоидные предшественники подвергаются ступенчатому созреванию, в ходе которого синтезируется пре-Т-клеточный рецептор (англ. T-cell receptor, TCR), стимуляция которого индуцирует коэкспрессию CD4 и CD8. СD4+CD8+ Т-клетки синтезируют зрелые рецепторы TCR/CD3 и взаимодействуют с эпителиальными клетками тимуса, экспрессирующими аутоантигены. Далее СD4+CD8+ Т-клетки проходят отрицательный отбор (гибель клеток из-за гиперстимуляции), отсроченный апоптоз (гибель клеток из-за недостаточной стимуляции) или положительный отбор (выживание клеток за счет адекватной стимуляции). Отобранные СD4+CD8+ Т-клетки затем мигрируют в мозговое вещество тимуса и далее дифференцируются в CD4 или CD8 Т-клетки. После отбора Т-клетки покидают тимус. Эта сложная серия событий организована несколькими факторами транскрипции (PU.1, Ikaros, Notch1, GATA3, TCF-1, E2A, HEB, BCL11b, Runx1/CBFβ, Klf2 и Foxo1), которые действуют на разных стадиях дифференцировки [10].
После созревания тимуса наивные CD4+ и CD8+ Т-клетки рециркулируют через кровеносную систему и лимфоидные органы, приобретая, таким образом, вполне зрелый фенотип. В частности, CD4-положительные лимфоциты могут далее дифференцироваться в отдельные регуляторные (Treg) или хелперные (Th) субпопуляции Т-клеток (Th1, Th2, Th17 и Tfh). Дифференциация этих популяций зависит от экспрессии ограниченного числа факторов транскрипции, известных как «мастер-регуляторы» дифференцировки Т-клеток (рис. 1) [11]. К ним относятся FOXP3 для Treg, Tbet для Th1, GATA3 для Th2, RORγt для Th17 и Bcl6 для Tfh. Экспрессия этих транскрипционных факторов регулируется специфическими цитокинами. В частности, воздействие интерферона γ и интерлейкина 12 (ИЛ-12) способствует экспрессии Tbet, в то время как высокие уровни ИЛ-4 индуцируют GATA3. ИЛ-6, ИЛ-10, ИЛ-2 и трансформирующий фактор роста β (англ. transforming growth factor β, TGFβ) усиливают экспрессию FOXP3, тогда как ИЛ-17, ИЛ-21, ИЛ-23, TGFβ и ИЛ-6 индуцируют RORγt. ИЛ-6 и ИЛ-21 стимулируют экспрессию Bcl6 и приобретение фенотипа Tfh [12].
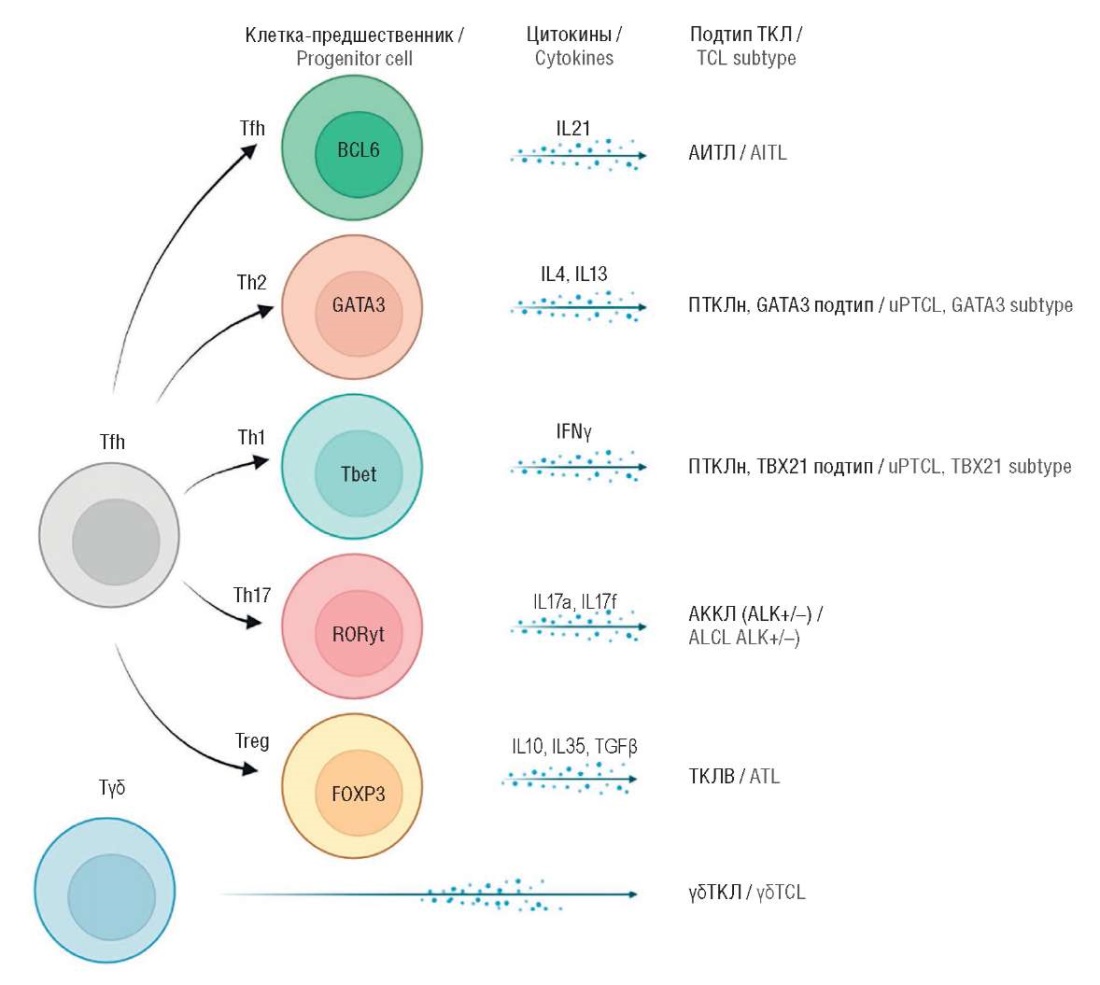
Рисунок 1. Транскрипционные факторы дифференцировки Т-клеток [11].
Tfh (англ. T follicular helpers) – фолликулярные Т-хелперы; IL (англ. interleukin) – интерлейкин; IFN (англ. interferon) – интерферон; TGF (англ. transforming growth factor) – трансформирующий фактор роста; ТКЛ – Т-клеточная лимфома; АИТЛ – ангиоиммунобластная Т-клеточная лимфома; ПТКЛн – периферическая Т-клеточная лимфома неспецифицированная; АККЛ – анапластическая крупноклеточная лимфома; ALK (англ. anaplastic lymphoma kinase) – киназа анапластической лимфомы; ТКЛВ – Т-клеточный лейкоз взрослых
Figure 1. Transcription factors of T-cell differentiation [11].
Tfh – T follicular helpers; IL – interleukin; IFN – interferon; TGF – transforming growth factor; TCL – T-cell lymphoma; AITL – angioimmunoblastic T-cell lymphoma; uPTCL – unspecified peripheral T-cell lymphoma; ALCL – anaplastic large cell lymphoma; ALK – anaplastic lymphoma kinase; ATL – adult T-cell leukemia
В течение многих лет «мастер-регуляторы» считались основными действующими лицами дифференцировки Th-клеток. Однако современная концепция гласит, что транскрипционным факторам отводится второстепенная роль, т.к. они, в свою очередь, могут изменять уровень своей экспрессии под действием факторов окружающей среды, таких как воспаление, инфицирование Т-лимфотропным вирусом человека 1-го типа (англ. human T-lymphotropic virus type 1, HTLV-1) или EBV, гипоксия и т.д. [13]. Именно такой интегрированный сценарий и обеспечивает молекулярную основу пластичности как нормальных, так и опухолевых Th-клеток [12].
В последние годы появляется все больше литературных данных о том, что глубокая реконструкции генома и транскриптома может помочь в понимании патогенеза ПТКЛ, постановке дифференциального диагноза, а также в определении прогноза и стратификации пациентов на ответчиков и неответчиков на некоторые виды терапии в онкологии [5].
J. Iqbal et al. с помощью методов секвенирования нового поколения (англ. next generation sequencing, NGS) предложили дополнительно классифицировать ПТКЛн на основании экспрессии генов [14]. Было обнаружено 3 подтипа: ПТКЛн с характеристиками хелперных Т-клеток типа 1, экспрессирующих Tbet (TBX21), ПТКЛ с характеристиками Th2, экспрессирующих GATA-связывающий белок 3 (GATA3), и ПТКЛн без четких признаков. У пациентов в подгруппе ПТКЛ-GATA3 прогноз был хуже, чем в подгруппе ПТКЛ-TBX21. Агрессивное клиническое течение в подгруппе ПТКЛ-GATA3, предположительно, связано с более высокой активностью онкогенных путей и более тяжелыми геномными аномалиями относительно ПТКЛ-TBX21 [14][15].
С. Amador et al. исследовали возможности иммуногистохимического (ИГХ) метода по воспроизведению классификации «клеток происхождения», первоначально предложенных с помощью методов NGS. На парафиновых срезах использовались антитела к GATA3 и TBX21 и их белкам-мишеням CCR4 и CXCR3. Проценты положительности опухолевых клеток с помощью иммунохимии и RNAseq высоко коррелировали, благодаря чему стало возможно создание ИГХ-алгоритма для молекулярной классификации ПТКЛн в клинической практике [6].
ПТКЛ являются гетерогенными, агрессивными НХЛ и обычно связаны с плохим прогнозом при традиционной химиотерапии. CHOP2 является наиболее часто назначаемой терапией первой линии при ПТКЛ. За исключением АККЛ, СНОР обеспечивает частоту общего ответа (англ. objective response rate, ORR) 60–80%, полный ответ (англ. сomplete response, CR) 30–40% и долгосрочную выживаемость, измеряемую 5-летней общей выживаемостью (англ. overall survival, OS), в диапазоне 20–30% [16–18]. Несмотря на то что трансплантация аутологичных стволовых клеток может удлинить выживаемость без прогрессирования (англ. рrogression-free survival, PFS) у некоторых пациентов, рецидивы остаются обычным явлением [17][19]. Разработка новых вариантов терапии первой линии ПТКЛ, а также поиск биомаркеров ответа и резистентности на новые виды терапии необходимы для улучшения качества и продолжительности ответа, а в конечном итоге – улучшения выживаемости.
ПТКЛ с Т-фолликулярным хелперным фенотипом (ПТКЛ-Tfh), включающим АИТЛ и подмножество вариантов ПТКЛн с Tfh, характеризуется повторяющимися мутациями в генах, которые выступают регуляторами процесса метилирования: Tet-метил-цитозиндиоксигеназа 2 (англ. ten-eleven translocation 2, TET2), изоцитратдегидрогеназа 2 (англ. isocitrate dehydrogenase 2, IDH2) и ДНК (цитозин-5)-метилтрансфераза 3A (англ. DNA methyltransferase 3A, DNMT3A) [20][21].
TET2 – это фермент, который превращает 5-метилцитозин в 5-гидроксиметилцитозин, обеспечивая обратимость процесса метилирования ДНК. IDH1/2 – фермент, катализирующий превращение изоцитрата в α-кетоглутарат. В результате мутаций в IDH1/2 фермент приобретает свойство катализировать превращение α-кетоглутарата в онкометаболит 2-гидроксиглутарат, который ингибирует TET, приводя к гиперметилированию [22]. DNMT – это ферменты, которые ковалентно присоединяют метильную группу к 5-С положению цитозина с образованием 5-метилцитозина. Основными представителями DNMT у человека являются DNMT1, TRDMT1, DNMT3a и DNMT3b.
В исследованиях на когортах пациентов с ПТКЛ показано, что интегративный анализ экспрессии генов и метилирования промоторов выявил рекуррентно гиперметилированные гены, участвующие в передаче сигналов TCR и дифференцировке Т-клеток, которые, вероятно, способствуют лимфомагенезу, что дает веские основания для клинического применения гипометилирующих агентов [23][24].
Азацитидин является эпигенетическим модификатором, который в низких дозах ингибирует ДНК-метилтрансферазу [25] и снижает метилирование ДНК. В более высоких дозах азацитидин, являясь аналогом нуклеозида цитидина, встраивается непосредственно в молекулы ДНК и РНК, обусловливая прямой цитотоксический эффект, приводящий к гибели клеток. Согласно данным литературы уровень метилирования ДНК в раковых клетках значительно снижен по всему геному [26]. Основной причиной снижения уровня метилирования в раковых клетках является деметилирование повторяющихся последовательностей участков генома [27]. Однако было показано, что в раковых клетках сосуществуют как низкие, так и высокие уровни метилирования ДНК. Низкие уровни метилирования ДНК связаны с активацией протоонкогенов, что приводит к геномной нестабильности, в то время как высокие уровни вызывают молчание промоторов генов-супрессоров опухолей, что приводит к инактивации последних [28]. Прием азацитидина в низкой дозе восстанавливает экспрессию генов-супрессоров и приводит к нормальной дифференцировке клеток.
Азацитидин проявляет клиническую активность в качестве монотерапии и в комбинации при рецидивирующей и рефрактерной ПТКЛ. Отдельный агент 5-азацитидин, вводимый подкожно в стандартной дозе 75 мг/м2 ежедневно в течение 7 дней каждые 28 дней, изучался в ретроспективном когортном исследовании 12 пациентов с рецидивирующей и рефрактерной АИТЛ. ORR составила 75% с CR на уровне 50% [29].
Пероральный азацитидин в комбинации с ромидепсином изучали в многоцентровом исследовании фазы I/II при рецидивирующей и рефрактерной ПТКЛ [30][31]. При максимальной переносимой дозе перорального азацитидина 300 мг в дни 1–14, а ромидепсина – 14 мг/м2 в дни 8, 15 и 22 каждые 35 дней эта эпигенетическая комбинация приводила к высокой частоте ответа, особенно при ПТКЛ-Tfh, где ORR составляла 80%, а CR – 67%. Эти результаты свидетельствуют о наследственно-специфической предрасположенности ПТКЛ к эпигенетически таргетной терапии. Возможность применения азацитидина в комбинации с ритуксимабом и CHOP была продемонстрирована в исследованиях фазы I при диффузной В-крупноклеточной лимфоме (ДВККЛ), в т.ч. в одном исследовании, которое показало, что пероральный прием азацитидина в дозе 300 мг в течение 14 дней можно безопасно сочетать со стандартной дозой R-CHOP [32][33].
Одной из часто встречающихся мутаций в ПТКЛ c Tfh-фенотипом является мутация в киназе RhoA. RhoA принадлежит к семейству Rho малых гуанозинтрифосфатаз, группе Ras-подобных белков, ответственных за регуляцию цитоскелета [34]. Однако в работе, где исследовалась связь данной мутации с ответом на азацитидин у пациентов с ПТКЛ, не было обнаружено достоверной связи [29].
Одним из перспективных биомаркеров ответа на азацитидин может быть клональность репертуаров TCR – уникальных последовательностей, соответствующих третьему гипервариабельному участку рецептора (англ. third complementarity determining region, CDR3). Одна CDR3-последовательность, уникальная по своему нуклеотидному составу, соответствует одному индивидуальному клону Т-клеток.
Репертуар Т-клеточных рецепторов в общем виде описывается двумя параметрами: разнообразием (общим количеством уникальных последовательностей CDR3 региона – чем больше количество уникальных последовательностей CDR3, тем выше разнообразие репертуара TCR) и клональностью (количеством прочтений, приходящихся на один клон Т-клеток – на один клон может приходиться много прочтений, и чем больше размер таких клонов относительно остальных вариантов, тем выше клональность репертуара; если же репертуар представлен клонами, приблизительно одинаковыми по размеру, его клональность считается низкой).
Т-клеточный рецептор состоит из двух разных полипептидных цепей (гетеродимер) – TCRα и TCRβ (либо TCRγ и TCRδ). Т-клетки с αβTCR называют αβТ-клетками, это наиболее распространенный и каноничный вариант Т-клеток. Второй вариант Т-клеток, с γδTCR, называют γδТ-клетками, это небольшая группа клеток со специфическими функциями и определенной локализацией в организме.
Анализ TCR проводится либо на данных RNAseq, когда из общей смеси отсеквенированных транскриптов с помощью специального софта (MiXCR) выбираются прочтения, соответствующие CDR3-последовательностям TCR, либо на данных таргетного секвенирования (англ. repertoire sequencing, RepSeq).
По данным литературы, ответ на гипометилирующие агенты может быть связан с уменьшением клональности TCR. Так, у пациентов с миелодиспластическим синдромом (МДС) при появлении новых клонотипов наблюдалась более высокая частота ответа на децитабин и азацитидин [35]. J. Ruan et al. также показали тенденцию к снижению клональности TCRB (p=–0,10) в ответ на праймирование азацитидином [36]. Это говорит о том, что ответ на терапию азацитидином может быть частично обусловлен иммунитетом, опосредованным Т-клетками, и что иммунная терапия, нацеленная на адаптивную иммунную систему, может играть значительную роль у отдельных пациентов с МДС. В работе J. Grimm et al. показана предиктивная роль сниженной клональности TCR до начала терапии азацитидином у пациентов с острым миелоидным лейкозом [37]. Больные с повышенным разнообразием T-клеточных репертуаров до лечения имели достоверное бо́льшие PFS и ОS.
Накопленный массив данных привел к инициации мультицентрового исследования по применению перорального азацитидина в качестве праймирования перед CHOP у пациентов с ранее не леченной ПТКЛ (NCT03542266). Всего в исследование был включен 21 пациент с ранее не леченной ПТКЛ, которые были зарегистрированы в 4 центрах с июня 2018 г. по март 2020 г. Двухлетняя OS составила 68,4% (95% доверительный интервал (ДИ) 47,3–89,4) для всех пациентов и 76,1% (95% ДИ 55,6–96,5%) для подгруппы ПТКЛ-Tfh.
Механизм действия азацитидинового прайминга был исследован с помощью интегративного геномного, транскриптомного и метиломного анализов. Наблюдалась активация генов, связанных с апоптозом и воспалением, включая интерферон типа I, фактор некроза опухоли α, а также макрофагов и сигналинга ИЛ-2. Благоприятная ассоциация мутаций TET2 с выживаемостью идентифицирует потенциальный целевой биомаркер восприимчивости, в то время как мутации DNMT3A связаны с потенциальным прогрессированием и резистентностью к лечению [36].
Гипометилирование продолжает исследоваться в межгрупповом рандомизированном исследовании фазы II ALLIANCE A051902, в котором сравнивают пероральный азацитидин в комбинации с CHOP/CHOEP, ингибитор фосфатидил-инозитол-3-киназы (англ. phosphoinositide-3-kinase, PI3K) дувелисиб в комбинации с CHOP/CHOEP со стандартной группой CHOP/CHOEP при CD30-отрицательной ПТКЛ (NCT04803201).
Среди новых нехимиотерапевтических препаратов бортезомиб и ромидепсин показали значительную активность при широком спектре лимфоидных злокачественных опухолей с хорошим профилем безопасности, отсутствием кумулятивной токсичности и принципиально иными механизмами действия, чем химиотерапия, которой первоначально подвергается большинство пациентов.
Среди ингибиторов гистонов деацетилазы (англ. histone deacetylases, HDAC) ромидепсин показал наиболее мощную клиническую активность при изучении аналогичных заболеваний. Среди ингибиторов протеасомы наибольший опыт применения у бортезомиба при лимфомах. Имеются сведения о комбинации ингибиторов HDAC и ингибиторов PI3K. Более того, доклинические данные показывают, что PI3K-ингибиторы могут оказывать синергетический эффект в индуцировании апоптоза при сочетании с HDAC-ингибиторами по сравнению с использованием одного из них в отдельности [38][39].
Синергизм между этими двумя классами препаратов был продемонстрирован в исследованиях in vitro на лимфоме Беркитта, немелкоклеточном раке легкого, эндометриальной саркоме, раке яичников, почечно-клеточной карциноме и хроническом миелогенном лейкозе [40–45]. В клеточных линиях кожной Т-клеточной лимфомы было показано, что вориностат сам по себе изменяет сигнализацию рецептора Т-клеток, MAPK- и JAK-STAT-пути, что приводит к ингибированию фосфорилирования AKT. Добавление ингибиторов PI3K приводит к синергическому цитотоксическому эффекту в этих клеточных линиях, что подтверждается тем фактом, что ингибиторы PI3K также влияют на путь mTOR/Akt [46].
Аналогичные результаты были получены и при других гематологических злокачественных опухолях. Кроме того, существуют доказательства синергизма между ингибиторами протеасомы и ингибиторами PI3K. В клеточных линиях фолликулярной лимфомы (ФЛ) и мантийно-клеточной лимфомы есть доказательства того, что ингибирование пути P13K/mTOR/Akt может реактивировать чувствительность к ингибиторам протеасомы, таким как бортезомиб [47][48].
Сигнальный путь PI3K/Akt/mTOR вовлечен в регуляцию основных процессов жизнедеятельности клетки, включая рост, пролиферацию и выживание. PI3K – это ключевой регуляторный белок данного пути. Семейство PI3K представлено тремя классами (I, II и III), которые отличаются по функциональной роли и субстратной специфичности (табл. 1).
Таблица 1. Гены, кодирующие фосфатидил-инозитол-3-киназу класса I
Table 1. Genes encoding phosphatidylinositol-3-kinase class I
|
Класс / Class |
Изоформа / Isoform |
Ген / Gene |
Преимущественная экспрессия / Predominant expression |
|
Ia |
α |
PIK3CA |
Повсеместная / Widespread |
|
Ia |
β |
PIK3CB |
|
|
Ia |
δ |
PI3KCD |
Гемопоэтические и иммунные клетки / Hemopoietic and immune cells |
|
Ib |
γ |
PIK3CG |
Молекула PI3K представляет собой гетеродимер и состоит из двух субъединиц: каталитической и регуляторной, каждая из которых кодируется разными генами. Класс PI3K I является наиболее изученным, т.к. участвует непосредственно в канцерогенезе, и в зависимости от типа субъединиц разделяется на подклассы IA и IB [49]. Класс IA представлен комплексом из каталитической субъединицы р110 (изоформы р110α, р110β, р110γ) и регуляторной субъединицы р85 (изоформы р85α, р55α, р50α, р85β и р55γ). Субъединицы p110α, p110β и p110δ кодируются генами PIK3CA, PIK3CB и PI3KCD соответственно. Класс IB представлен геном PIK3CG, который кодирует каталитическую субъединицу p110γ. Изоформы p110α и p110β экспрессируются во всех тканях организма, в то время как экспрессия p110δ и p110γ ограничена преимущественно лейкоцитами и гемопоэтическими клетками [50].
Функциональная роль PI3K класса II менее изучена, однако есть исследования, свидетельствующие о возможном участии в регуляции клеточного роста и ангиогенеза [51]. Киназы класса III отвечают преимущественно за регуляцию внутриклеточного транспорта везикул и белков [52].
Дувелисиб является селективным двойным ингибитором каталитических субъединиц p110γ и p110δ фермента PI3K. Конкурентно замещая молекулы аденозинтрифосфата (АТФ) в АТФ-связывающих «карманах» каталитических субъединиц, препарат вызывает полную инактивацию фермента.
В дополнение к важности PI3K-δ/γ в функционировании Т-клеток [53–55] необходимо учитывать изменения опухолевого микроокружения, вызванные ингибированием PI3K-δ/PI3K-γ. В некоторых подтипах ПТКЛ (особенно в АИТЛ) молекулярное профилирование позволило выявить признаки микроокружения опухоли, связанные в дальнейшем с плохим исходом [5].
Ингибиторы PI3K-δ и PI3K-γ активно изучаются в гематологических злокачественных новообразованиях. Иделалисиб, ингибитор PI3K-δ, был одобрен Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (англ. U.S. Food and Drug Administration, FDA) для использования при рецидивирующей ФЛ и хроническом лимфолейкозе (ХЛЛ) на основании рандомизированного плацебо-контролируемого исследования, которое продемонстрировало общую частоту ответа 81% и улучшение общей выживаемости на 12 мес [56]. Исследования дувелисиба in vitro показали ингибирование роста клеточных линий ХЛЛ, ДВККЛ, а также T-клеточного острого лимфобластного лейкоза [57][58].
Исследования фазы I дувелисиба проводились у пациентов как с индолентной НХЛ, так и с ТКЛ. В исследование по ТКЛ вошли 33 пациента, и было обнаружено, что максимально переносимая доза составляет 75 мг перорально 2 раза в день. У поддающихся оценке пациентов общая частота ответа составила 42% (47% при ПТКЛ и 38% при ТКЛК). Среднее время ответа составило 1,9 мес. У 26 (79%) пациентов отмечены нежелательные явления ≥3 ст., наиболее частыми из которых были повышение активности аланинаминотранферазы (АЛТ) и аспартатаминотрансферазы (АСТ) (n=12; 36%), сыпь (n=7; 21%) и нейтропения (n=5; 15%); 30% пациентов прекратили лечение из-за побочных реакций [59]. В фазе I исследования пациентов с индолентными НХЛ было обнаружено, что наиболее частыми побочными эффектами ≥3 ст., возникшими на фоне лечения, были повышение активности АЛТ/АСТ (n=13; 41%) и диарея (n=7; 22%) [60].
Бортезомиб – первый в своем классе ингибитор протеасомы, который обратимо связывается с треониновым остатком протеасомы 26S. Протеасома 26S присутствует в ядре и цитозоле всех эукариотических клеток и является ключевым компонентом, катализирующим расщепление основных белков, которые участвуют в управлении жизненным циклом клеток. Бортезомиб ингибирует химотрипсиноподобное действие протеасомы, вызывает торможение протеолиза и приводит к апоптозу.
Ингибиторы протеасом первоначально были одобрены для использования при рецидивирующей и/или рефрактерной множественной миеломе, и впоследствии было обнаружено, что они эффективны при мантийно-клеточных лимфомах, а также при индолентных В-клеточных лимфомах [61]. При ФЛ бортезомиб в комбинации с ритуксимабом дает 55–70% ответа [62]. Кроме того, при ранее не леченной ДВККЛ у пациентов, получавших R-CHOP + бортезомиб, наблюдался 100% ответ [63].
Бортезомиб коммерчески доступен с мая 2003 г. Он был одобрен FDA для использования при множественной миеломе, лимфоме из мантийных клеток и внесен в список Национальной комплексной онкологической сети (англ. National Comprehensive Cancer Network, NCCN) для использования при ТКЛ, системном амилоидозе легких цепей и макроглобулинемии Вальденстрема [64]. Известно, что бортезомиб блокирует активацию ядерного фактора каппа B (англ. nuclear factor kappa B, NF-κB), предотвращая деградацию IκB в клеточных линиях множественной миеломы [65].
Наиболее распространенные проявления токсичности бортезомиба включают диарею, запор, утомляемость, периферическую невропатию, тромбоцитопению и анемию. Исследования показали, что частота периферической невропатии значительно снижается при подкожном введении по сравнению с внутривенным введением без влияния на эффективность [66].
Бортезомиб также продемонстрировал эффективность при ранее леченных ТКЛК с частотой ответа 67% в качестве монотерапии. Ответы на бортезомиб у этих пациентов были стойкими и продолжались от 7 до 14 мес в исследовании фазы II [67]. Кроме того, фаза I исследования CHOP с бортезомибом в качестве терапии первой линии для агрессивных ТКЛ продемонстрировала частоту ответа 61% [68]. Исследование фазы II CHOP с бортезомибом в качестве терапии первой линии при агрессивных ТКЛ показало общий уровень ответа 87% и 74% у пациентов с полным ответом с диагнозами ПТКЛн, АИТЛ и АККЛ [69]. Таким образом, ингибиторы протеасом, в т.ч. бортезомиб, являются перспективными для применения в качестве комбинации при рефрактерной и рецидивирующей ПТКЛ.
Ромидепсин является ингибитором HDAC, которая катализирует удаление ацетильной группы из остатков лизина белка (включая гистоновые и транскрипционные факторы). Ингибирование HDAC приводит к накоплению ацетильных групп, что приводит к изменениям в структуре хроматина и активации фактора транскрипции, что вызывает прекращение роста клеток (остановку клеточного цикла в фазах G1 и G2/M) и их гибель. Предполагаемый механизм действия в В-клеточных лимфомах осуществляется через протоонкоген BCL6, который является высокоактивным транскрипционным репрессором в зародышевой В-клетке. Нарушенная активность BCL6 приводит к подавлению генов, участвующих в активации лимфоцитов, дифференцировке, остановке клеточного цикла и апоптозу. Более того, известно, что накопление ацетилированного BCL6 снижает транскрипционную репрессивную функцию, способствуя транскрипции полезных генов и накоплению проапоптотических продуктов в злокачественной клетке [70].
Такие ингибиторы HDAC, как вориностат, белиностат и ромидепсин, были одобрены FDA для использования в качестве монотерапии при рецидивирующих/рефрактерных ТКЛ. Ромидепсин был оценен в исследовании GPI-04-0001 и NCI Study 1312 на 135 испытуемых с ТКЛК [71]. Общая частота ответа составила 41% (55/135), а частота полного ответа – 7% (10/135). Ромидепсин был активен во всех очагах заболевания, включая кожу, лимфатические узлы, внутренности и кровь. Эти данные привели к одобрению FDA препарата ромидепсин для лечения ТКЛК в сентябре 2009 г. Активность ромидепсина при ПТКЛ была подтверждена в многоцентровом исследовании фазы II с участием 131 пациента с рецидивом ПТКЛ [72]. Частота объективного ответа составила 25% (33/130), включая 15% (19/130) с полным ответом. Наиболее распространенными нежелательными явлениями класса ≥3 были тромбоцитопения (24%), нейтропения (20%) и инфекции (все типы, 19%). На основе этих данных было получено одобрение FDA препарата ромидепсин для лечения ПТКЛ в июне 2011 г.
Учитывая потребность в лучшей переносимости и более активной терапии для пациентов с рецидивирующими и рефрактерными Т-клеточными злокачественными опухолями, было инициировано параллельное исследование с использованием дувелисиба в комбинации с бортезомибом или ромидепсином (NCT02783625). Добавление ромидепсина к дувелисибу оказалось безопасным и снижало количество побочных реакций по сравнению с монотерапией дувелисибом. Общий ответ составил 55% (35/64), а полный – 34% (22/64). Частый последующий переход к аллотрансплантации подтверждает ценность комбинации дувелисиба с ромидепсином для пациентов с рефрактерной ПТКЛ. По данным биомаркерного анализа, проведенного в рамках клинического испытания, показано, что мутации TET2 были предикторами ответа, в то время как мутации TP53 наблюдались исключительно у неответчиков [73].
Многие современные подходы к лечению рефрактерной ПТКЛ ведут пациентов по пути многократного комбинированного лечения цитотоксическими препаратами. В результате, как правило, возникают ограничивающие лечение токсические эффекты, а повторные курсы цитотоксической химиотерапии демонстрируют все меньшую отдачу. Сохраняется значительная потребность в более активных методах лечения и комбинациях, при этом существует множество новых подходов, исследующих режимы, которые меняют парадигму на долгосрочное или поддерживающее лечение с использованием препаратов без кумулятивной токсичности. Не менее важным является исследование возможностей персонализированного назначения препаратов. В представленной работе рассмотрены перспективы персонализации назначения таких противоопухолевых препаратов, как азацитидин, дувелисиб, ромидепсин и бортезомиб, для терапии рефрактерной или рецидивирующей ПТКЛ.
1. ALK (англ. anaplastic lymphoma kinase) – киназа анапластической лимфомы.
2. CHOP – циклофосфамид (англ. Cyclophosphamide), гидроксидаунорубицин (англ. Hydroxydaunorubicin), онковин (англ. Oncovin), преднизон (англ. Prednisone).
1. Vose J., Armitage J., Weisenburger D. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol. 2008; 26 (25): 4124–30. https://doi.org/10.1200/JCO.2008.16.4558.
2. Alaggio R., Amador C., Anagnostopoulos I., et al. The 5th edition of the World Health Organization Classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. 2022; 36 (7): 1720–48. https://doi.org/10.1038/s41375-022-01620-2.
3. A clinical evaluation of the International Lymphoma Study Group classification of non-Hodgkin’s lymphoma. The Non-Hodgkin's Lymphoma Classification Project. Blood. 1997; 89 (11): 3909–18.
4. Cheson B.D., Fisher R.I., Barrington S.F., et al. Recommendations for initial evaluation, staging, and response assessment of Hodgkin and non-Hodgkin lymphoma: the Lugano classification. J Clin Oncol. 2014; 32 (27): 3059–3068. https://doi.org/10.1200/JCO.2013.54.8800.
5. Iqbal J., Weisenburger D.D., Greiner T.C., et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010; 115 (5): 1026–36. https://doi.org/10.1182/blood-2009-06-227579.
6. Amador C., Greiner T.C., Heavican T.B., et al. Reproducing the molecular subclassification of peripheral T-cell lymphoma-NOS by immunohistochemistry. Blood. 2019; 134 (24): 2159–70. https://doi.org/10.1182/blood.2019000779.
7. Торшин И.Ю., Громова О.А., Тетруашвили Н.К. Хемотранскриптомный анализ синергизма D-хироинозитола и миоинозитола в контексте постгеномной фармакологии. Акушерство и гинекология. 2022; 9: 135–45. https://doi.org/10.18565/aig.2022.9.135-145.
8. Лила А.М., Торшин И.Ю., Громов А.Н. и др. Фармакоинформационные исследования хондропротекторов. Современная ревматология. 2021; 15 (5): 114–20. https://doi.org/10.14412/1996-7012-2021-5-114-120.
9. Громова О.А., Торшин И.Ю., Сорокин А.И. и др. Хемотранскриптомный анализ молекулы этилметилгидроксипиридина сукцината в контексте постгеномной фармакологии. Неврология, нейропсихиатрия, психосоматика. 2020; 12 (5): 130–7. https://doi.org/10.14412/2074-2711-2020-5-130-137.
10. Inghirami G., Chan W.C., Pileri S. Peripheral T-cell and NK cell lymphoproliferative disorders: cell of origin, clinical and pathological implications. Immunol Rev. 2015; 263 (1): 124–59. https://doi.org/10.1111/imr.12248.
11. Marchi E., O’Connor O.A. The rapidly changing landscape in mature T-cell lymphoma (MTCL) biology and management. CA Cancer J Clin. 2020; 70 (1): 47–70. https://doi.org/10.3322/caac.21589.
12. Pizzi M., Margolskee E., Inghirami G. Pathogenesis of peripheral T cell lymphoma. Annu Rev Pathol. 2018; 13: 293–320. https://doi.org/10.1146/annurev-pathol-020117-043821.
13. Josefowicz S.Z. Regulators of chromatin state and transcription in CD4 T-cell polarization. Immunology. 2013; 139 (3): 299–308. https://doi.org/10.1111/imm.12115.
14. Iqbal J., Wright G., Wang C., et al. Gene expression signatures delineate biological and prognostic subgroups in peripheral T-cell lymphoma. Blood. 2014; 123 (19): 2915–23. https://doi.org/10.1182/blood-2013-11-536359.
15. Wang T., Feldman A.L., Wada D.A., et al. GATA-3 expression identifies a high-risk subset of PTCL, NOS with distinct molecular and clinical features. Blood. 2014; 123 (19): 3007–15. https://doi.org/10.1182/blood-2013-12-544809.
16. Mourad N., Mounier N., Brière J., et al. Clinical, biologic, and pathologic features in 157 patients with angioimmunoblastic T-cell lymphoma treated within the Groupe d’Etude des Lymphomes de l'Adulte (GELA) Trials. Blood. 2008; 111 (9): 4463–70. https://doi.org/10.1182/blood-2007-08-105759.
17. Reimer P., Rüdiger T., Geissinger E., et al. Autologous stem-cell transplantation as first-line therapy in peripheral T-cell lymphomas: results of a prospective multicenter study. J Clin Oncol. 2009; 27 (1): 106–13. https://doi.org/10.1200/JCO.2008.17.4870.
18. Simon A., Peoch M., Casassus P., et al. Upfront VIP-reinforced-ABVD (VIP-rABVD) is not superior to CHOP/21 in newly diagnosed peripheral T cell lymphoma. Results of the randomized phase III trial GOELAMS-LTP95. Br J Haematol. 2010; 151 (2): 159–66. https://doi.org/10.1111/j.1365-2141.2010.08329.x.
19. d’Amore F., Relander T., Lauritzsen G.F., et al. Up-front autologous stem-cell transplantation in peripheral T-cell lymphoma: NLG-T-01. J Clin Oncol. 2012; 30 (25): 3093–9. https://doi.org/10.1200/JCO.2011.40.2719.
20. Swerdlow S.H., Campo E., Pileri S.A., et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016; 127 (20): 2375–90. https://doi.org/10.1182/blood-2016-01-643569.
21. de Leval L., Rickman D.S., Thielen C., et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007; 109 (11): 4952–63. https://doi.org/10.1182/blood-2006-10-055145.
22. Waitkus M.S., Diplas B.H., Yan H. Biological role and therapeutic potential of IDH mutations in cancer. Cancer Cell. 2018; 34 (2): 186–95. https://doi.org/10.1016/j.ccell.2018.04.011.
23. Wang C., McKeithan T.W., Gong Q., et al. IDH2R172 mutations define a unique subgroup of patients with angioimmunoblastic T-cell lymphoma. Blood. 2015; 126 (15): 1741–52. https://doi.org/10.1182/blood-2015-05-644591.
24. Odejide O., Weigert O., Lane A.A., et al. A targeted mutational landscape of angioimmunoblastic T-cell lymphoma. Blood. 2014; 123 (9): 1293–6. https://doi.org/10.1182/blood-2013-10-531509.
25. Dan H., Zhang S., Zhou Y., Guan Q. DNA methyltransferase inhibitors: catalysts for antitumour immune responses. Onco Targets Ther. 2019; 12: 10903–16. https://doi.org/10.2147/OTT.S217767.
26. Feinberg A.P., Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004; 4 (2): 143–53. https://doi.org/10.1038/nrc1279.
27. Yoder J.A., Walsh C.P., Bestor T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997; 13 (8): 335–40. https://doi.org/10.1016/s0168-9525(97)01181-5.
28. Akhavan-Niaki H., Samadani A.A. DNA methylation and cancer development: molecular mechanism. Cell Biochem Biophys. 2013; 67 (2): 501–13. https://doi.org/10.1007/s12013-013-9555-2.
29. Lemonnier F., Dupuis J., Sujobert P., et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood. 2018; 132 (21): 2305–9. https://doi.org/10.1182/blood-2018-04-840538.
30. O’Connor O.A., Falchi L., Lue J.K., et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: a multicenter phase 1 study. Blood. 2019; 134 (17): 1395–405. https://doi.org/10.1182/blood.2019001285.
31. Falchi L., Ma H., Klein S., et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood. 2021; 137 (16): 2161–70. https://doi.org/10.1182/blood.2020009004.
32. Clozel T., Yang S., Elstrom R.L., et al. Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 2013; 3 (9): 1002–19. https://doi.org/10.1158/2159-8290.CD-13-0117.
33. Martin P., Bartlett N.L., Chavez J.C., et al. Phase 1 study of oral azacitidine (CC-486) plus R-CHOP in previously untreated intermediate- to high-risk DLBCL. Blood. 2022; 139 (8): 1147–59. https://doi.org/10.1182/blood.2021011679.
34. Palomero T., Couronné L., Khiabanian H., et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014; 46 (2): 166–70. https://doi.org/10.1038/ng.2873.
35. Abbas H.A., Reville P.K., Jiang X., et al. Response to hypomethylating agents in myelodysplastic syndrome is associated with emergence of novel TCR clonotypes. Front Immunol. 2021; 12: 659625. https://doi.org/10.3389/fimmu.2021.659625.
36. Ruan J., Moskowitz A.J., Mehta-Shah N., et al. Multicenter phase 2 study of oral azacitidine (CC-486) plus CHOP as initial treatment for peripheral T-cell lymphoma. Blood. 2023; blood.2022018254. https://doi.org/10.1182/blood.2022018254.
37. Grimm J., Simnica D., Jäkel N., et al. Azacitidine-induced reconstitution of the bone marrow T cell repertoire is associated with superior survival in AML patients. Blood Cancer J. 2022; 12 (1): 19. https://doi.org/10.1038/s41408-022-00615-7.
38. Bodo J., Zhao X., Sharma A., et al. The phosphatidylinositol 3-kinases (PI3K) inhibitor GS-1101 synergistically potentiates histone deacetylase inhibitor-induced proliferation inhibition and apoptosis through the inactivation of PI3K and extracellular signal-regulated kinase pathways. Br J Haematol. 2013; 163 (1): 72–80. https://doi.org/10.1111/bjh.12498.
39. Bodo J., Zhao X., Sharma A., et al. The PI3K inhibitor GS-1101 (CAL-101) synergistically potentiates HDAC-induced proliferation inhibition and apoptosis through the activation of JNK in lymphoma cells. Blood. 2012; 120 (21): 3714. https://doi.org/10.1182/blood.V120.21.3714.3714.
40. Ozaki K.I., Kosugi M., Baba N., et al. Blockade of the ERK or PI3K-Akt signaling pathway enhances the cytotoxicity of histone deacetylase inhibitors in tumor cells resistant to gefitinib or imatinib. Biochem Biophys Res Commun. 2010; 391 (4): 1610–5. https://doi.org/10.1016/j.bbrc.2009.12.086.
41. Zhou C., Qiu L., Sun Y., et al. Inhibition of EGFR/PI3K/AKT cell survival pathway promotes TSA’s effect on cell death and migration in human ovarian cancer cells. Int J Oncol. 2006; 29 (1): 269–78.
42. Quan P., Moinfar F., Kufferath I., et al. Effects of targeting endometrial stromal sarcoma cells via histone deacetylase and PI3K/AKT/mTOR signaling. Anticancer Res. 2014; 34 (6): 2883–97.
43. Ferreira A.C., Robaina M.C., Rezende L.M., et al. Histone deacetylase inhibitor prevents cell growth in Burkitt’s lymphoma by regulating PI3K/Akt pathways and leads to upregulation of miR-143, miR-145, and miR-101. Ann Hematol. 2014; 93 (6): 983–93. https://doi.org/10.1007/s00277-014-2021-4.
44. Yamada T., Horinaka M., Shinnoh M., et al. A novel HDAC inhibitor OBP-801 and a PI3K inhibitor LY294002 synergistically induce apoptosis via the suppression of survivin and XIAP in renal cell carcinoma. Int J Oncol. 2013; 43 (4): 1080–6. https://doi.org/10.3892/ijo.2013.2042.
45. Nguyen T., Dai Y., Attkisson E., et al. HDAC inhibitors potentiate the activity of the BCR/ABL kinase inhibitor KW-2449 in imatinib-sensitive or -resistant BCR/ABL+ leukemia cells in vitro and in vivo. Clin Cancer Res. 2011; 17 (10): 3219–32. https://doi.org/10.1158/1078-0432.CCR-11-0234.
46. Wozniak M.B., Villuendas R., Bischoff J.R., et al. Vorinostat interferes with the signaling transduction pathway of T-cell receptor and synergizes with phosphoinositide-3 kinase inhibitors in cutaneous T-cell lymphoma. Haematologica. 2010; 95 (4): 613–21. https://doi.org/10.3324/haematol.2009.013870.
47. Bhende P.M., Park S.I., Lim M.S., et al. The dual PI3K/mTOR inhibitor, NVP-BEZ235, is efficacious against follicular lymphoma. Leukemia. 2010; 24 (10): 1781–4. https://doi.org/10.1038/leu.2010.154.
48. Kim A., Park S., Lee J.E., et al. The dual PI3K and mTOR inhibitor NVP-BEZ235 exhibits anti-proliferative activity and overcomes bortezomib resistance in mantle cell lymphoma cells. Leuk Res. 2012; 36 (7): 912–20. https://doi.org/10.1016/j.leukres.2012.02.010.
49. Alzahrani A.S. PI3K/Akt/mTOR inhibitors in cancer: at the bench and bedside. Semin Cancer Biol. 2019; 59: 125–32. https://doi.org/10.1016/j.semcancer.2019.07.009.
50. Li H., Prever L., Hirsch E., Gulluni F. Targeting PI3K/AKT/mTOR signaling pathway in breast cancer. Cancers (Basel). 2021; 13 (14): 3517. https://doi.org/10.3390/cancers13143517.
51. Yoshioka K., Yoshida K., Cui H., et al. Endothelial PI3K-C2α, a class II PI3K, has an essential role in angiogenesis and vascular barrier function. Nat Med. 2012; 18 (10): 1560–9. https://doi.org/10.1038/nm.2928.
52. Raiborg C., Schink K.O., Stenmark H. Class III phosphatidylinositol 3-kinase and its catalytic product PtdIns3P in regulation of endocytic membrane traffic. FEBS J. 2013; 280 (12): 2730–42. https://doi.org/10.1111/febs.12116.
53. Reif K., Okkenhaug K., Sasaki T., et al. Cutting edge: differential roles for phosphoinositide 3-kinases, p110γ and p110δ, in lymphocyte chemotaxis and homing. J Immunol. 2004; 173 (4): 2236–40. https://doi.org/10.4049/jimmunol.173.4.2236.
54. Soond D.R., Bjørgo E., Moltu K., et al. PI3K p110δ regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. 2010; 115 (11): 2203–13. https://doi.org/10.1182/blood-2009-07-232330.
55. Okkenhaug K., Patton D.T., Bilancio A., et al. The p110δ isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol. 2006; 177 (8): 5122–8. https://doi.org/10.4049/jimmunol.177.8.5122.
56. Furman R.R., Sharman J.P., Coutre S.E., et al. Idelalisib and rituximab in relapsed chronic lymphocytic leukemia. N Engl J Med. 2014; 370 (11): 997–1007. https://doi.org/10.1056/NEJMoa1315226.
57. Balakrishnan K., Peluso M., Fu M., et al. Inhibition of PI3K-δ and -γ isoforms by IPI-145 in chronic lymphocytic leukemia overcomes signals from PI3K/AKT/S6 pathway and promotes apoptosis. Blood. 2013; 122 (21): 4167. https://doi.org/10.1182/blood.V122.21.4167.4167.
58. Huang X., Proctor J., Yang Y., et al. The potent PI3K-δ,γ inhibitor, IPI-145, exhibits preclinical activity in murine and human T-cell acute lymphoblastic leukemia. Blood. 2013; 122 (21): 1438. https://doi.org/10.1182/blood.V122.21.1438.1438.
59. Horwitz S.M., Porcu P., Flinn I., et al. Duvelisib (IPI-145), a phosphoinositide-3-kinase-δ,γ inhibitor, shows activity in patients with relapsed/refractory T-cell lymphoma. Blood. 2014; 124 (21): 803. https://doi.org/10.1182/blood.V124.21.803.803.
60. Flinn I., Oki Y., Patel M., et al. A Phase 1 evaluation of duvelisib (IPI-145), a PI3K-δ,γ inhibitor, in patients with relapsed/refractory iNHL. Blood. 2014; 124 (21): 802. https://doi.org/10.1182/blood.V124.21.802.802.
61. Goy A., Younes A., McLaughlin P., et al. Phase II study of proteasome inhibitor bortezomib in relapsed or refractory B-cell non-Hodgkin’s lymphoma. J Clin Oncol. 2005; 23 (4): 667–75. https://doi.org/10.1200/JCO.2005.03.108.
62. Zinzani P.L., Khuageva N.K., Wang H., et al. Bortezomib plus rituximab versus rituximab in patients with high-risk, relapsed, rituximab-naïve or rituximab-sensitive follicular lymphoma: subgroup analysis of a randomized phase 3 trial. J Hematol Oncol. 2012; 5: 67. https://doi.org/10.1186/1756-8722-5-67.
63. Ruan J., Martin P., Furman R.R., et al. Bortezomib plus CHOP-rituximab for previously untreated diffuse large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol. 2011; 29 (6): 690–7. https://doi.org/10.1200/JCO.2010.31.1142.
64. Anderson K.C., Alsina M., Bensinger W., et al. Waldenström’s macroglobulinemia/lymphoplasmacytic lymphoma, version 2.2013. J Natl Compr Canc Netw. 2012; 10 (10): 1211–9. https://doi.org/10.6004/jnccn.2012.0128.
65. Karin M., Cao Y., Greten F.R., Li Z.W. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002; 2 (4): 301–10. https://doi.org/10.1038/nrc780.
66. Moreau P., Pylypenko H., Grosicki S., et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol. 2011; 12 (5): 431–40. https://doi.org/10.1016/S1470-2045(11)70081-X.
67. Zinzani P.L., Musuraca G., Tani M., et al. Phase II trial of proteasome inhibitor bortezomib in patients with relapsed or refractory cutaneous T-cell lymphoma. J Clin Oncol. 2007; 25 (27): 4293–7. https://doi.org/10.1200/JCO.2007.11.4207.
68. Lee J., Suh C., Kang H.J., et al. Phase I study of proteasome inhibitor bortezomib plus CHOP in patients with advanced, aggressive T-cell or NK/T-cell lymphoma. Ann Oncol. 2008; 19 (12): 2079–83. https://doi.org/10.1093/annonc/mdn431.
69. Kim S.J., Yoon D.H., Kang H.J., et al. Bortezomib in combination with CHOP as first-line treatment for patients with stage III/IV peripheral T-cell lymphomas: a multicentre, single-arm, phase 2 trial. Eur J Cancer. 2012; 48 (17): 3223–31. https://doi.org/10.1016/j.ejca.2012.06.003.
70. Hatzi K., Melnick A. Breaking bad in the germinal center: how deregulation of BCL6 contributes to lymphomagenesis. Trends Mol Med. 2014; 20 (6): 343–52. https://doi.org/10.1016/j.molmed.2014.03.001.
71. Rasheed W., Bishton M., Johnstone R.W., Prince H.M. Histone deacetylase inhibitors in lymphoma and solid malignancies. Expert Rev Anticancer Ther. 2008; 8 (3): 413–32. https://doi.org/10.1586/14737140.8.3.413.
72. Coiffier B., Pro B., Prince H.M., et al. Results from a pivotal, open-label, phase II study of romidepsin in relapsed or refractory peripheral T-cell lymphoma after prior systemic therapy. J Clin Oncol. 2012; 30 (6): 631–6. https://doi.org/10.1200/JCO.2011.37.4223.
73. Lavrol Clin. Cancer trial results. Horwitz S., Nikitina A., Kotlov N., et al. The combination of duvelisib and romidepsin (DR) is highly active against relapsed/refractory peripheral T-cell lymphoma with low rates of transaminitis: final results and biomarker analysis. 2021. URL: https://clin.larvol.com/abstract-detail/ASH%202021/52318948 (дата обращения 10.02.2023).
Сорокина Мария Андреевна – аспирант кафедры фармакологии ФГБОУ ВО «Ивановская государственная медицинская академия» Минздрава России, аналитик Нейрокампуса-2030 ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России.
Scopus Author ID: 57226747037; РИНЦ SPIN-код: 4142-8679.
Шереметевский пр-т, 8, Иваново 153012
ул. Островитянова, 1, Москва 117997
Рахтеенко Арина Владимировна – врач-педиатр отделения трансплантации гемопоэтических стволовых клеток № 1 ФГБУ
ул. Саморы Машела, 1, Москва 117997
Гришина Татьяна Романовна – д.м.н., профессор, заведующая кафедрой фармакологии
РИНЦ SPIN-код: 1241-0701.
Шереметевский пр-т, 8, Иваново 153012
Сорокина М.А., Рахтеенко А.В., Гришина Т.Р. Новые фармакотерапевтические подходы к лечению периферической Т-клеточной лимфомы. ФАРМАКОЭКОНОМИКА. Современная фармакоэкономика и фармакоэпидемиология. 2023;16(2):291-302. https://doi.org/10.17749/2070-4909/farmakoekonomika.2023.170
Sorokina M.А., Rakhteenko A.V., Grishina T.R. New pharmacotherapeutic approaches for the treatment of peripheral T-cell lymphoma. FARMAKOEKONOMIKA. Modern Pharmacoeconomics and Pharmacoepidemiology. 2023;16(2):291-302. (In Russ.) https://doi.org/10.17749/2070-4909/farmakoekonomika.2023.170

Издатель: ООО ИРБИС
Адрес: 101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Телефон: +7 (495) 649 54 95
Email: alexandra.moskvicheva@irbis-1.ru


































